T cell immunity and immunosuppression in cancer
The immune system has a powerful ability to recognize and kill cancer cells, but its function is often suppressed within tumours, preventing clearance of disease. Understanding how tumors suppress immunity has led to major therapeutic advances, including checkpoint inhibitors targeting PD-1 and CTLA-4. However, most patients do not durably respond to today’s immunotherapies.
Our laboratory aims to discover how cancers suppress T cell responses to evade destruction and resist immunotherapy. We aim to translate fundamental discoveries into new more effective therapies for patients with cancer, combining mechanistic immunology with cutting-edge approaches including CRISPR-based functional genetics and directed tumor evolution. Our research is increasingly focused on developing novel biologic, cellular and molecular therapies to improve treatment of advanced cancer patients or to prevent cancer recurrence in those at high risk of metastasis.
Suppression of T cell immunity within tumours
Tumors create hostile cellular and chemical environments that suppress anti-tumor immunity.
Regulatory T (Treg) cells suppress conventional T cell responses and are a major barrier to anti-tumor immunity (Figure 1). We are interested in understanding how Treg cells develop and function. We discovered that the transcriptional repressor BACH2 is essential for Treg cell development and established a widely-accepted molecular model for BACH2 function in lymphocytes (Roychoudhuri et al., Nature 2013; Roychoudhuri et al., Nature Immunology 2016). This work explained why genetic variations at the human BACH2 locus confer susceptibility to autoimmunity and allergy, and led to identification—through international collaboration—of BACH2-related Immunodeficiency and Autoimmunity (BRIDA), a previously unrecognised human monogenic disease, improving clinical management for affected patients worldwide (Afzali et al., Nature Immunology 2017). We showed how a distal enhancer at the prominent 11q13.5 autoimmune disease risk locus restricts gut inflammation by promoting expression of the TGF-β docking receptor GARP on Treg cells (Nasrallah et al., Nature 2020).
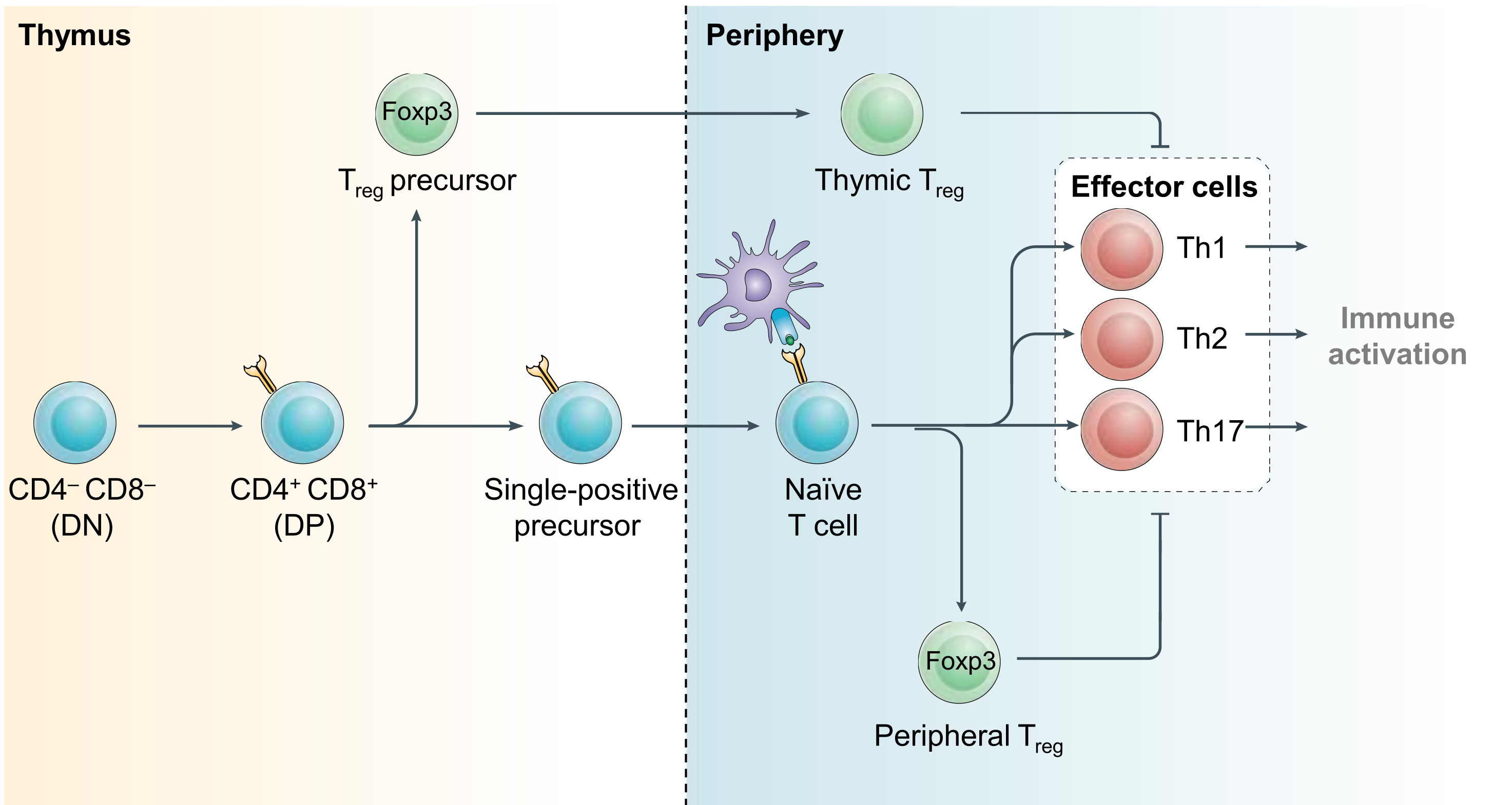
Figure 1. Regulatory T cell development. CD4+ effector and regulatory T (Treg) cells arise from common precursor cells within the thymus and periphery by exert opposing functions. Treg-mediated restraint of effector cell function is a critical immunoregulatory mechanism required to prevent lethal inflammation. Modified from Igarashi, Kurosaki and Roychoudhuri, Nat Rev Immunol 2017.
Within tumors, we discovered that activated Treg responses are maintained by quiescent stem-like progenitor cells, raising questions about how Treg populations are maintained within tumours (Grant et al., J Exp Med 2020). We identified CCR8 as a marker of highly suppressive tumor-infiltrating Treg cells and a distinct suppressive subset of conventional CD4+ T cells that mediate immunotherapy resistance through IL-10 production (Whiteside et al., Science Immunology 2023). We are now examining how therapeutic strategies could reprogram Treg cells within tumors, including OX40/CD137 bispecific antibodies that convert Tregs into IFN-γ-producing cells that enhance anti-tumor immunity (Imianowski et al., Cancer Res Commun 2024).
We are interested in identifying and targeting mechanisms by which the tumor microenvironment disables T cell responses. We discovered that dying tumor cells release intracellular potassium that suppresses T cell activation (Eil et al., Nature 2016) and that tumor acidity inactivates interleukin-2, enabling us to engineer pH-selective IL-2 variant with preferential activity in acidic tumors (Gaggero et al., Science Immunology 2022). We are now applying directed tumor evolution and high-throughput CRISPR screens to identify additional immunoregulatory mechanisms within tumors, with the goal of developing next-generation biological and small molecule approaches to enhance T cell function.
T Cell Maintenance and Dysfunction
Effective immune responses to cancer require T cells to persist over long periods while retaining their ability to kill tumor cells. We have studied the molecular programs that control T cell longevity and how tumors induce T cell dysfunction—insights we are now aim to translate into improved cell therapies for cancer patients (Figure 2).
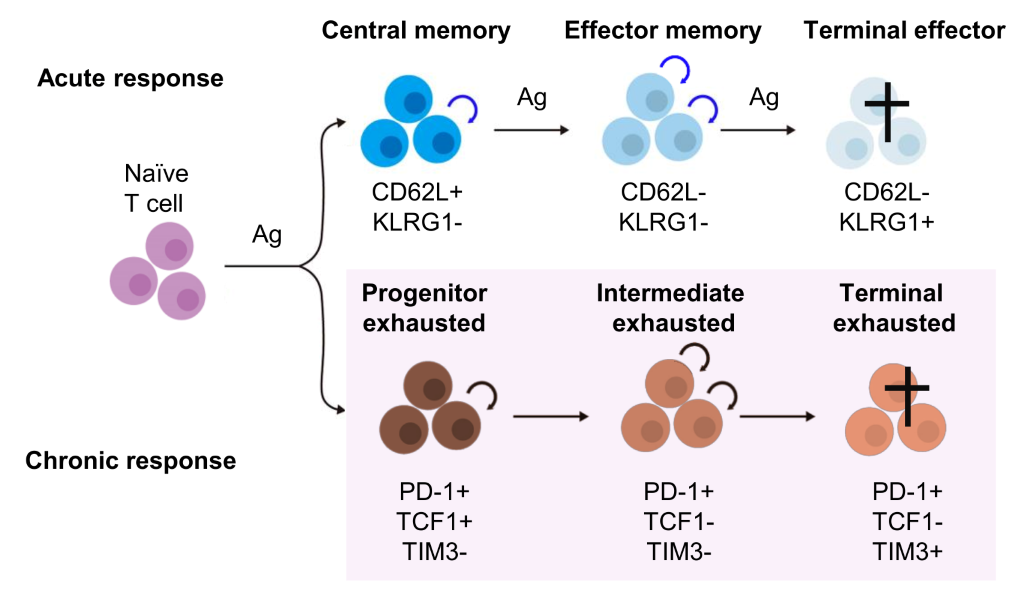
Figure 2. Maintenance of acute and chronic T cell responses. In both acute and chronic immune responses, T cells undergo progressive differentiation accompanied by acquisition of effector functions and loss of maintenance potential self-renewal and multipotency. This loss of maintenance potential is a key feature of the progression of T cells from naive, effector, and memory T cell subsets in acute responses, as well as progenitor, intermediate, and terminally exhausted T cells in chronic responses.
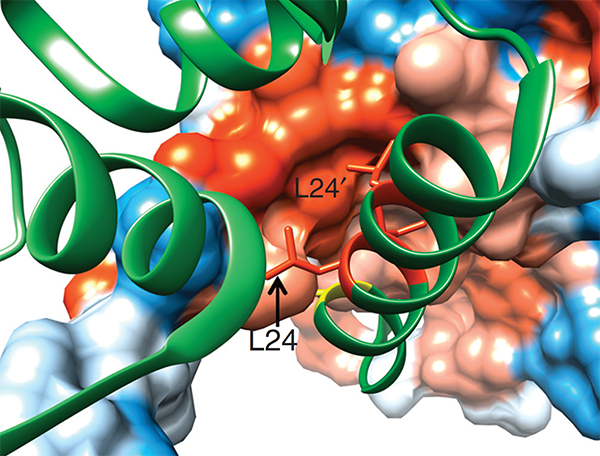
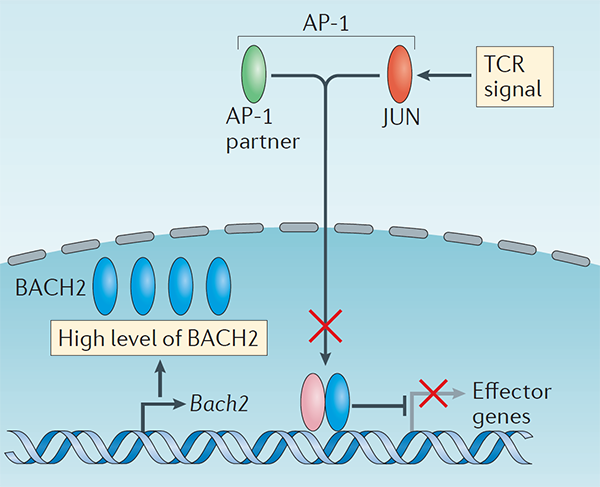
We conducted one of the earliest multiplexed single-cell gene expression analyses of immune cells, revealing heterogeneity in memory CD8+ T cell responses to vaccination (Flatz, Roychoudhuri et al., PNAS 2011) and defining the transcriptional and epigenetic programs of vaccine-induced memory T cells (Roychoudhuri et al., Vaccine 2015). We discovered that BACH2 functions as a regulator of T cell quiescence, promoting long-lived memory CD8+ T cell responses to viral infection (Roychoudhuri et al., Nature Immunology 2016). Mechanistically, BACH2 represses T cell receptor-driven effector programs by occupying AP-1 binding sites and blocking access by AP-1 transcription factors. This quiescence program extends beyond CD8+ T cells: we showed that long-term Treg cell maintenance depends on a quiescent subset marked by high Bach2 expression (Grant et al., J Exp Med 2020), and that BACH2 similarly constrains NK cell maturation, restricting NK-mediated immunosurveillance of lung metastasis (Imianowski et al., J Exp Med 2022). We contributed to studies showing that AKT pathway inhibition generates T cells with enhanced memory characteristics and superior anti-tumor activity in adoptive cell therapy (Crompton et al., Cancer Res 2015), and that memory T cell-driven differentiation can impair therapeutic efficacy (Klebanoff et al., J Clin Invest 2015).
We are now using these insights to engineer improved CAR T cell therapies. Multiple ongoing projects aim to harness T cell maintenance programs to generate cell products with enhanced persistence and anti-tumor function for patients with advanced cancers.
Immune Interception of Early Cancer and Cancer Metastasis
Advanced cancers are protected by a well-established immunosuppressive microenvironment that limits the efficacy of current immunotherapies. Yet early pre-malignant cancers and newly established or dormant micrometastases lack the highly immunosuppressive microenvironment of established tumours (Fig. 3). This creates an opportunity for therapies that utilize the immune system to prevent cancer metastasis in patients with early-stage cancer at risk of recurrence, or to prevent early cancer in high-risk individuals.
We found that aspirin prevents cancer metastasis by blocking platelet-derived thromboxane A2 (TXA2), which suppresses T cell immunity (Yang et al., Nature 2025). This work provides a mechanistic understanding of aspirin’s previously observed anti-metastatic activity. Building on this interest, we are developing preventative immunotherapies—including novel biologics and small molecules—to prevent cancer or its recurrence in high-risk patients. By targeting cancer before widespread metastatic dissemination occurs, we aim to improve outcomes for patients facing the greatest cancer burden.
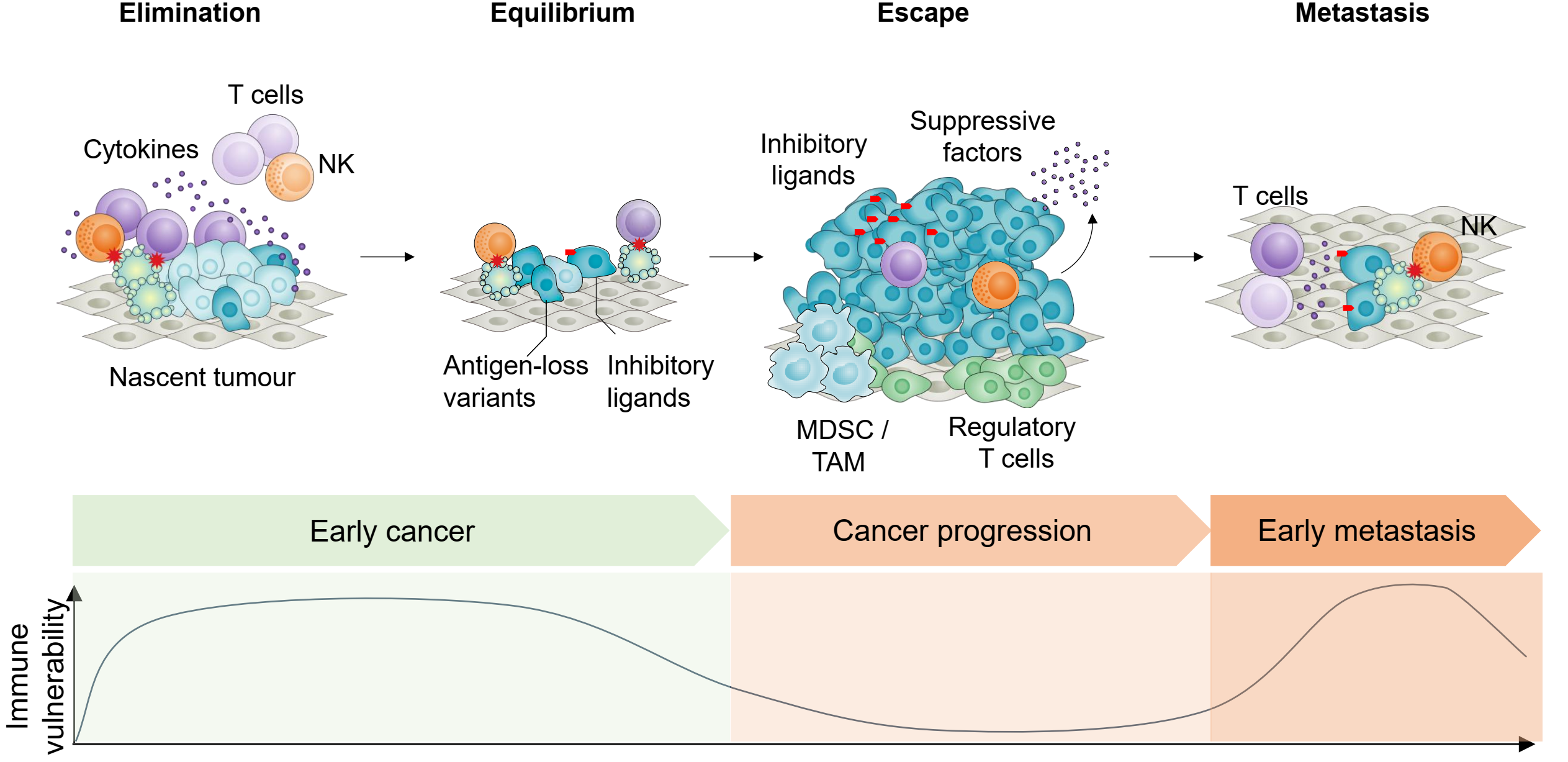
Figure 3. Immune vulnerability of cancer cells at distinct stages of cancer development provides a potential window for immune prevention. Cancer progression through elimination, equilibrium, and escape phases results in the establishment of a protective immunosuppressive microenvironment, featuring regulatory T cells, MDSCs, TAMs, and suppressive factors that shield advanced tumours from immune attack. However, early-stage cancers and disseminated micrometastases lack this immunosuppressive armor, remaining vulnerable to elimination by T cells and NK cells. The immune vulnerability curve (bottom) reveals two critical therapeutic windows: early cancers before escape mechanisms develop, and micrometastatic deposits that are deprived of the protective tumour microenvironment. This vulnerability of early and disseminated disease represents a major opportunity for preventive immunotherapy—targeting cancer when it is most susceptible to immune elimination rather than after immunosuppressive defenses are established.
We showed that T cells sense high oxygen concentrations in lung tissue through prolyl hydroxylase proteins, establishing an immunologically tolerant niche vulnerable to metastasis (Clever et al., Cell 2016). We demonstrated that NK cell maturation states determine metastatic susceptibility, with BACH2 limiting NK-mediated immunosurveillance of lung metastases (Imianowski et al., J Exp Med 2022).
We welcome inquiries from funders interested in supporting our fundamental and translational cancer immunology research and from talented scientists seeking to join our team.
Research Highlights
(For a full list of publications see below)

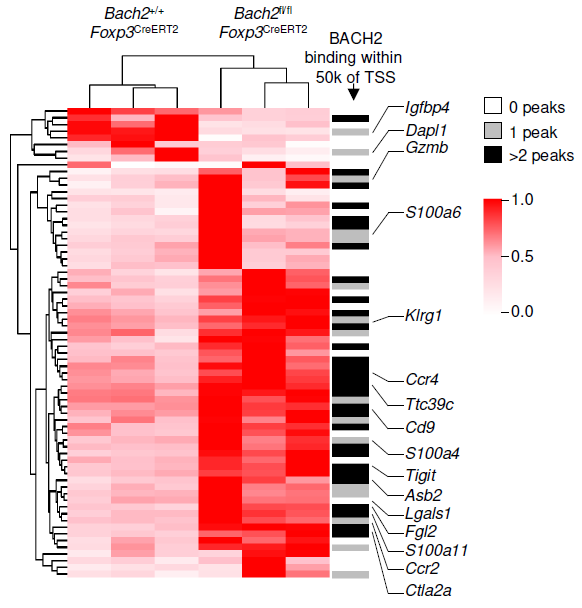
Adoptive T cell therapies are limited by poor persistence of transferred cells. We show that quantitative control of BACH2 dosage regulates differentiation along the stem-effector continuum, enabling engineering of synthetic CD8⁺ T cell states with persistent antitumor activity. While high-level BACH2 overexpression enforces quiescence and hinders tumor control, low-dose expression promotes persistence without compromising effector function by partially attenuating terminal differentiation programs. One of three papers published back-to-back showing that BACH2 can be exploited therapeutically for cell therapy by adjusting its dosage and timing.
Conti, Evans, von Linde et al., Nat Immunol (2026) [PDF]
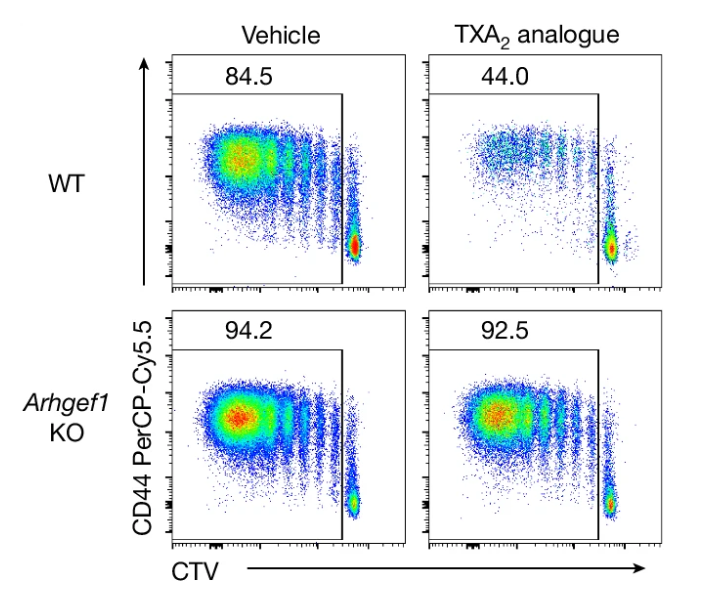
Retrospective clinical studies suggest that people who take aspirin may be at reduced risk of cancer metastasis. Here we show that aspirin enhances immunity to cancer metastasis by releasing T cells from suppression by platelet thromboxane A2 (TXA2). TXA2 acts on T cells to trigger an immunosuppressive pathway dependent on a guanine exchange factor ARHGEF1. Restricting the availability of TXA2 using aspirin or COX-1 inhibitors reduces the rate of metastasis in a manner dependent on T cell-intrinsic expression of ARHGEF1.
Yang, et al., Nature (2025) [PDF]
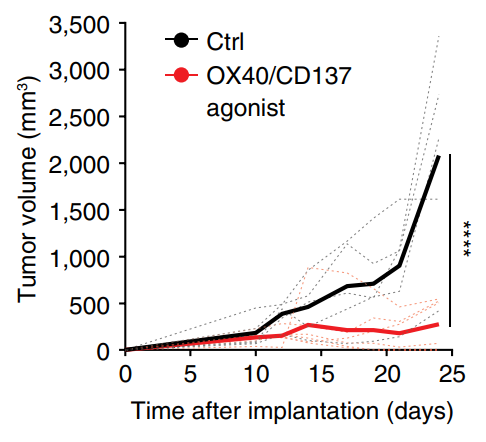
Tregs suppress immune responses to cancer. There is interest in reprogramming Tregs to contribute to antitumor immunity. Here, we show that OX40/CD137 bispecific agonists induce potent antitumor immunity in large part dependent upon functional reprogramming of Tregs to produce IFN-γ. Conditional deletion of Ifng in Foxp3+ Tregs and their progeny partially reversed the antitumor efficacy of OX40/CD137 bispecific agonist therapy, revealing that reprogramming of Tregs into IFNγ-producing cells contributes to the anti-tumor efficacy of OX40/CD137 bispecific agonists.
Imianowski, Kuo, et al., Cancer Res Commun (2024) [PDF]
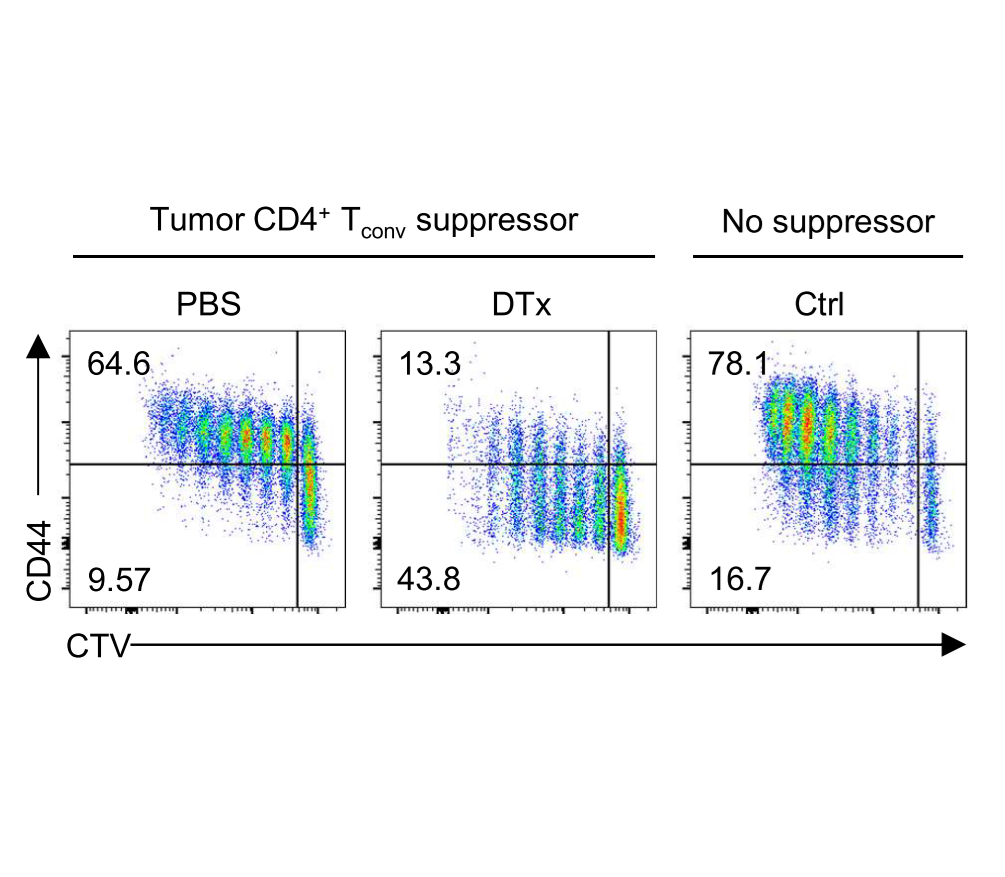
Strategies to disrupt Treg cell–mediated cancer immunosuppression have been met with limited clinical success. By modeling Treg cell–targeted immunotherapy in mice, we find that CD4+ Foxp3− conventional T (Tconv) cells acquire suppressive function limiting therapeutic efficacy. This activity is attributable to a Th2-like subset of CCR8+ Tconv cells which mediate IL-10–dependent suppression of antitumor immunity, revealing a counterregulatory layer of immunosuppression released upon therapeutic Treg cell depletion.
Whiteside, et al., Sci Immunol (2023) [PDF]
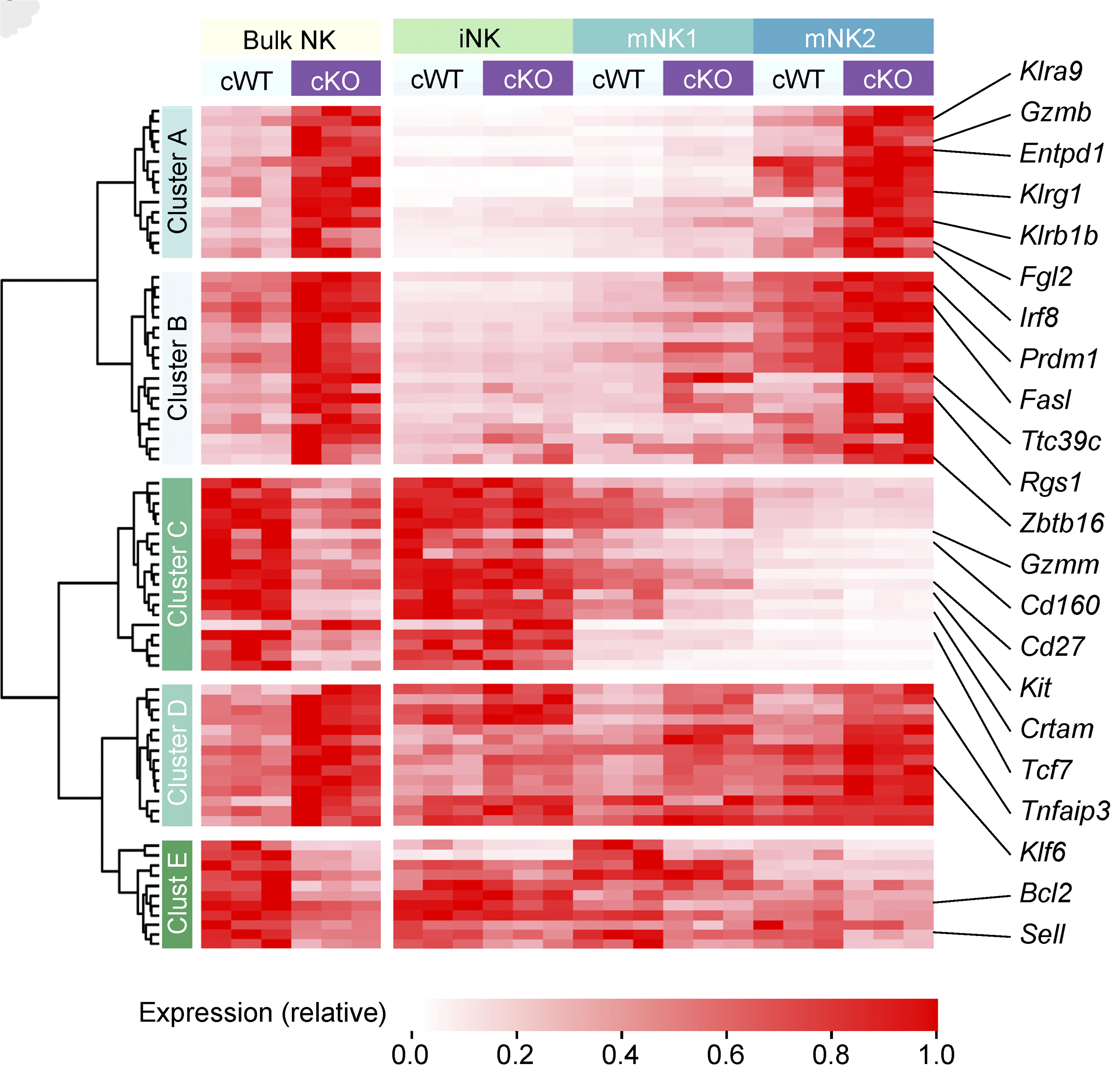
Natural killer (NK) cells are critical to immune surveillance against infections and cancer. Here we show that the transcription factor BACH2 functions as a critical negative regulator of NK cell function within tissues, restricting immunity to cancer metastasis. The findings show that the maturation state of NK cells in tissues is a major determinant of anti-metastatic immunity.
Imianowski, et al., J Exp Med (2022) [PDF]

Here, we show that IL-2, a cytokine critical to supporting CD8 T cell and NK cell functions within tumours exhibits markedly compromised binding to the IL-2Rα receptor chain in acidic extracellular environments. Directed evolution was used to screen for IL-2 variants with mutations at the receptor interface that enhanced receptor binding at low pH. A mutated “Switch-2” variant of IL-2 with strong receptor binding at pH 6 but minimal interaction at neutral pH exhibited enhanced in vivo antitumor activity with reduced toxicity in normal tissues.
Gaggero, Martinez-Fabregas, et al., Sci Immunol (2022) [PDF]
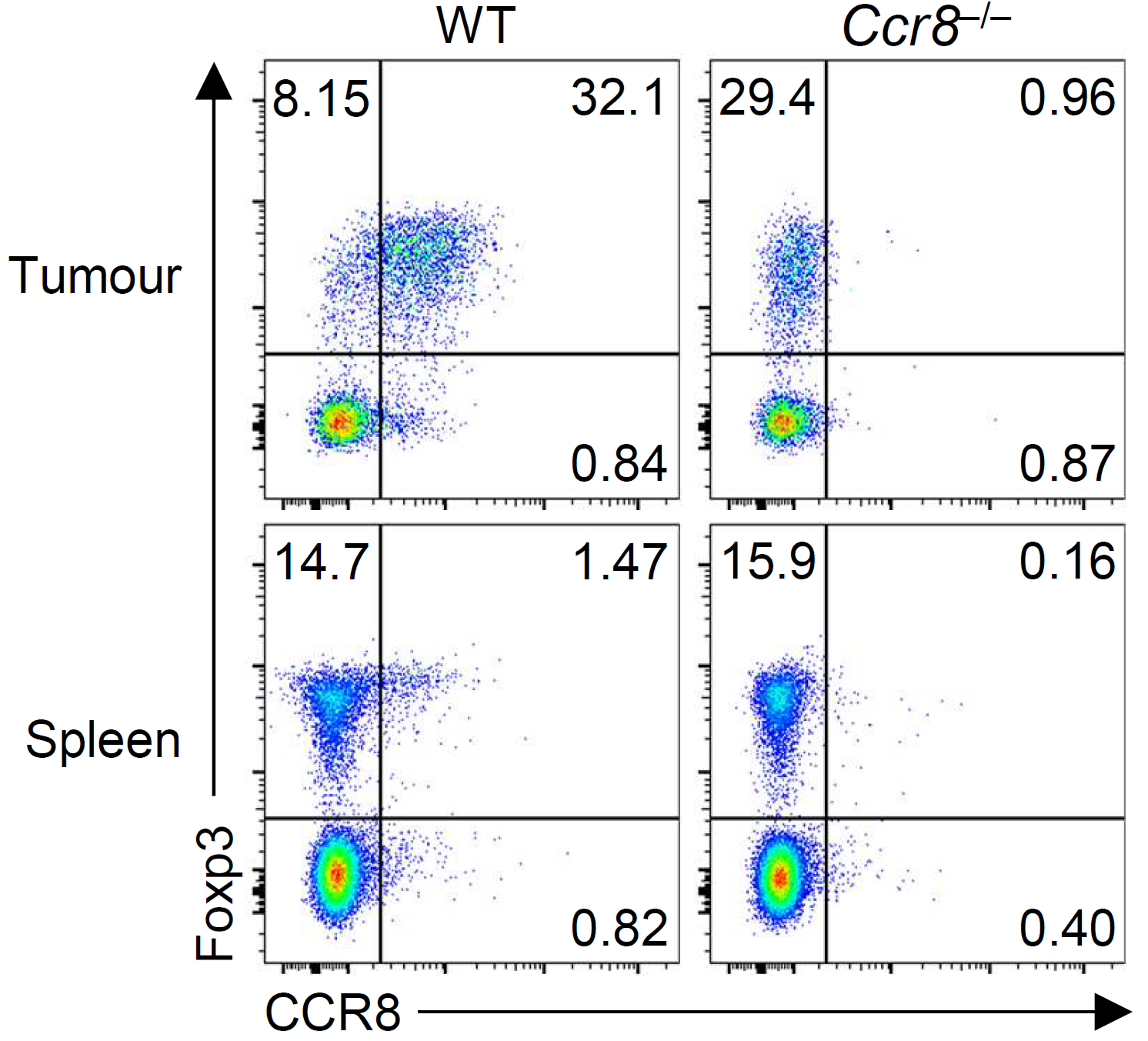
There is interest in selectively targeting the suppressive function of Treg cells within tumours. We show that high levels of CCR8 expression distinguish highly suppressive Treg cells within tumours, but that genetic deletion of CCR8 has minimal impact on Treg suppressive function or tumor immunity. These findings suggest that CCR8 is not required for Treg suppression within tumours and that depletion of CCR8+ Treg cells rather than blockade of CCR8 function will enable selective cancer immunotherapy.
Whiteside, et al., Immunology (2021) [PDF]
Genetic variations underlying risk of complex immune-mediated diseases are concentrated within the non-coding genome at enhancers. The functions of a vast majority of enhancers are unresolved. Using CRISPR-based mutagenesis of human disease-associated enhancer homologs in mice, we show that a distal enhancer at the human 11q13.5 immune disease risk locus promotes Treg-mediated suppression of colitis by driving expression of the TGF-β docking receptor GARP on Treg cells.
Nasrallah, Imianowski, et al., Nature (2020) [PDF]
Recommended by F1000; Commentary in Sci Immunol 5,eabe0976
Treg cells are maintained over long periods of time despite a continuous requirement for their suppressive function. This study established a critical function of quiescent cells in Treg maintenance. The transcription factor BACH2 is critical for Treg cell development. We show that in mature Treg cells, Bach2 is highly expressed in quiescent cells where it binds to enhancers of genes associated with Treg activation and represses their TCR- and AP-1-driven induction. By imposing quiescence in this subset of Treg cells, BACH2 promotes maintenance of Treg responses, and durable immune suppression in cancer.
Grant, Yang et al., J Exp Med (2020) [PDF]
This study describes a human monogenic disease resulting from inactivating mutations of BACH2, termed BACH2-related Immunodeficiency and Autoimmunity (BRIDA). Affected patients have lymphocyte-differentiation defects associated with immunodeficiency and inflammation of the intestinal tract and lungs. This work resulted from an international collaboration with clinical and academic researchers in the US and UK.
Afzali et al., Nat Immunol (2017) 18:813-823. [PDF]
Here we review roles of BACH family transcriptional repressors in innate and adaptive immunity. We highlight similarities at a molecular level in the cell-type-specific activities of the BACH factors, proposing that competitive interactions of BACH proteins with transcriptional activators of the bZIP family form a common mechanistic theme underlying their diverse actions.
Igarashi, Kurosaki and Roychoudhuri, Nat Rev Immunol (2017) 17:437-450. [PDF]
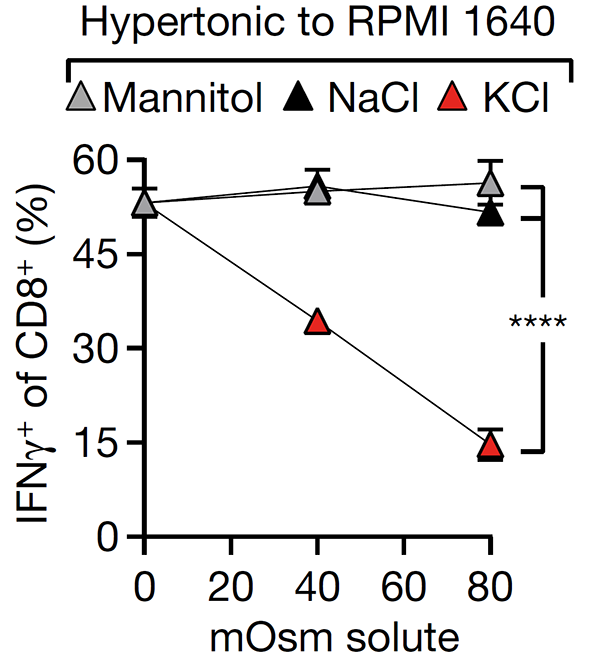
This study revealed that cell death in the tumour microenvironment releases intracellular potassium into the extracellular microenvironment of tumours, causing profound suppression of T cell activation. High extracellular potassium concentrations impair T cell receptor (TCR)-driven Akt-mTOR phosphorylation and effector programmes. Engineering CD8+ T cells to be resistant to high extracellular potassium in tumours improves adoptive immunotherapy.
Eil et al., Nature (2016) 537:539-543. [PDF]
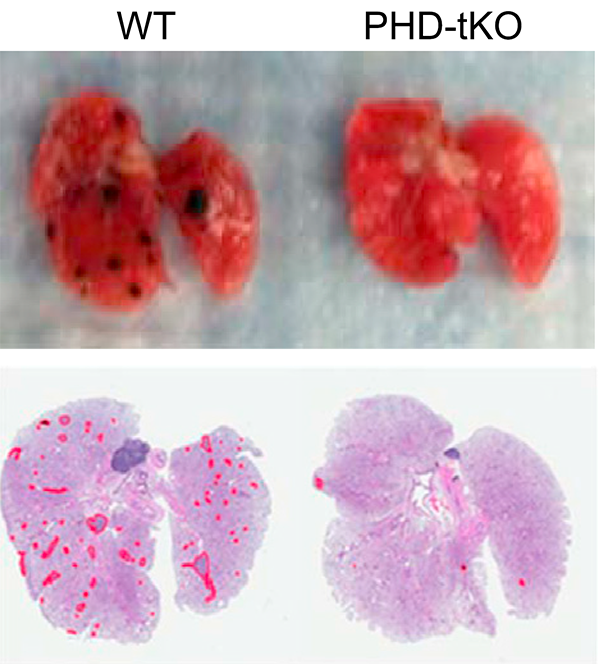
The lung is a frequent site of cancer metastasis but whether there is an immunological component to this susceptibility has been unclear. Here, we show that T cell responses to metastasis within the lung are restricted by high oxygen concentrations. The HIF prolyl hydroxylases PHD1, PHD2 and PHD3 mediate sensitivity of T cells to oxygen, which suppresses Th1 responses and promotes iTreg responses within the lung. As a consequence, immunologic permissivity of the lung to cancer metastasis is dependent upon PHD1, PHD2 and PHD3.
Clever et al., Cell (2016) 166:1117-1131 [PDF]
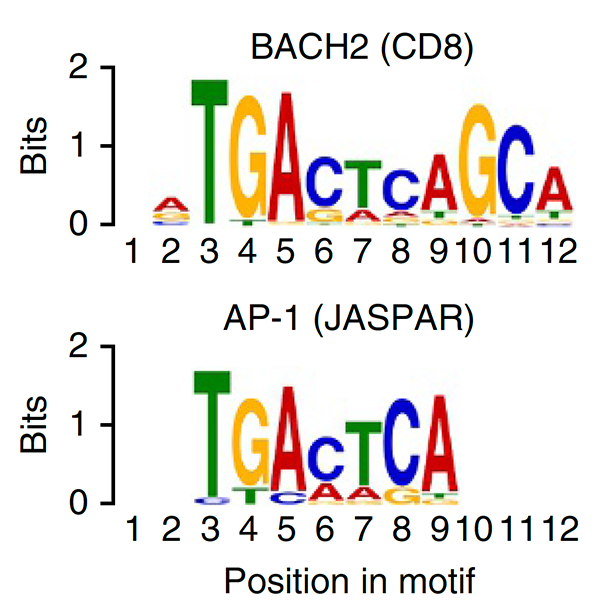
This study was the first to define BACH2 key regulator of lymphocyte quiescence. We found that BACH2 restrains effector differentiation to promote differentiation of long-lived memory CD8+ T cells after viral infection. We found that BACH2 binds to enhancers where it engages in steric competition with AP-1 factors, limiting TCR-driven induction of AP-1 target genes to promote memory formation. Subsequent work has shown that BACH2 is expressed in stem-like progenitors within T cell responses to cancer.
Roychoudhuri et al., Nat Immunol (2016) 17:851-860. [PDF]
Commentary by Sidwell and Kallies (Nat Immunol 17:744-5.
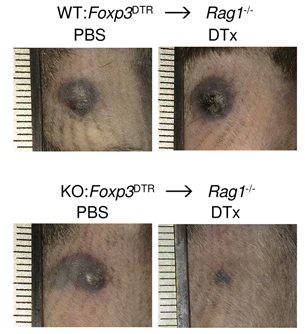
This study established a role of BACH2 in Treg-mediated tumour immunosuppression. This function is corroborated by results of a large in vivo screen of host restriction factors to tumour metastasis (Van der Weyden et al., Nature 2017) and provides a basis for a CRUK-funded drug development programme with CRUK Therapeutic Discovery Laboratories to identify small molecule inhibitors of the suppressive activity of BACH2.
Roychoudhuri et al., J Clin Invest (2016) 126:599-604. [PDF]
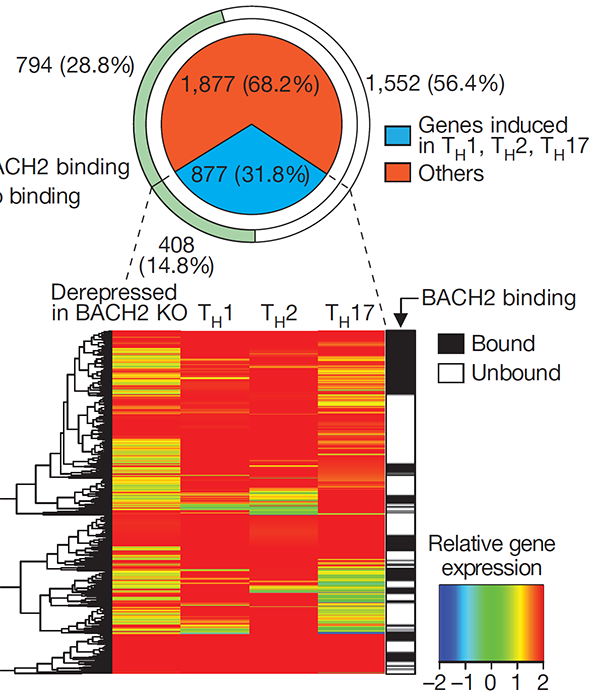
This study established the critical function of the transcription factor BACH2 in immunological tolerance. We found that BACH2 is required for early Treg cell development and its deletion in mice results in lethal inflammation. The findings contributed to our understanding of why genetic polymorphisms within the BACH2 locus in humans are associated with susceptibility to autoimmune and allergic diseases (reviewed in Igarashi, Kurosaki and Roychoudhuri, Nat Rev Immunol 2017).
Roychoudhuri et al., Nature (2013)</b> 498:506-10. [PDF]
Recommended by F1000
Group Members
[Team] [Join us]@RoychoudhuriLab
Collaborators:
- Klaus Okkenhaug (Pathology)
- David Adams (Sanger)
- Adrian Liston (Babraham)
- Enrico Lugli (Humanitas, Milan)
- Tim Halim (CRUK CI)
- Suman Mitra (INSERM Lille)
- Gosia Trynka (Sanger)
Environment:
- Department of Pathology (Cambridge)
- CRUK Cambridge Centre (Cambridge)
- Babraham Institute (Cambridge)

