T cell immunity and immunosuppression in cancer
T cells drive immune activation and promote clearance of infections and cancer. However, their function can provoke autoimmune and allergic inflammation. The immune system therefore employs a variety of suppressive mechanisms, known as immunoregulatory mechanisms, to restrain excessive T cell activation and prevent autoimmune and allergic inflammation. It is now known that such suppressive mechanisms inhibit anti-tumour immunity to drive deleterious immunosuppression in cancer. Immunoregulatory mechanisms therefore function as ‘brakes’ within the immune system and are important therapeutic targets in cancer. This is exemplified by the clinical efficacy of cancer immunotherapies targeting the immune ‘checkpoints’ PD-1 and CTLA-4 in certain cancers.
We believe that fundamental discovery in the fields of immune regulation and cancer immunosuppression will enable development of new and more effective therapies for patients with presently incurable autoimmune and allergic diseases and cancer. Our research falls within three key areas described below.
Suppression of T cell immunity in cancer
Cancers adapt to their immune environment to evade attack. By the time cancers are clinically relevant, they have learned to subvert the biochemical, metabolic and ionic environment of tumours, and co-opt the immunosuppressive functions of a variety of immunoregulatory cells, to drive immune dysfunction (Figure 1).
The laboratory is interested in gaining a mechanistic understanding of how T cell and NK cell function is impaired in cancer. We are applying directed tumour evolution and high-throughput CRISPR-based functional genetics, to identify novel immunoregulatory mechanisms operating within the tumour microenvironment.
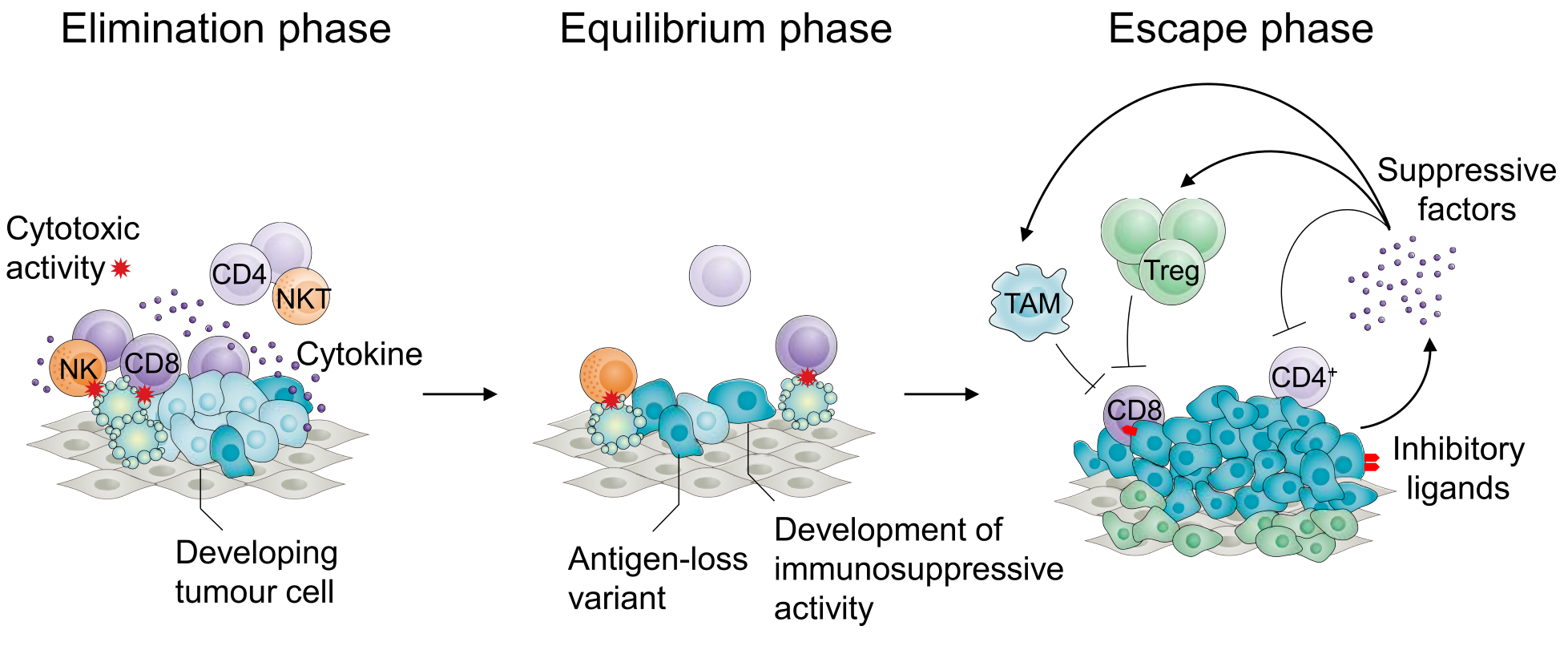
Figure 1. Phases of tumour development according to the cancer immunoediting hypothesis. Tumour development is characterized by an initial ‘elimination’ phase, during which a majority of cancer cells are destroyed by a variety of components of the innate and adaptive immune systems, including CD8+ T cells and NK cells. This process results, referred to as immunoediting, results in an ‘equilibrium’ phase, during which pressure from the immune system contributes to selection of tumour variants that do not express antigens targeted by the adaptive immune system or have developed mechanisms to suppress immune function. This gives rise to the ‘escape’ phase characterized by recruitment and support of the differentiation and proliferation of immunosuppressive cell types including Treg cells, tumour-associated macrophages and myeloid-derived suppressor cells, expression of inhibitory ligands and such as PD-L1 and production of immunosuppressive factors such as TGF-b resulting in evasion from immune control and unrestrained tumour growth.
Cancer immunosuppression by regulatory T (Treg) cells
Regulatory T (Treg) cells are rare immune cells with powerful suppressive functions. Loss of CD4+ Foxp3+ Treg cells results in lethal inflammation, while defects in their function are associated with autoimmunity and allergy. Treg cells powerfully suppress immune responses in cancer. There is intense medical interest in disrupting the immunosuppressive functions of Treg cells in cancer. However, efforts to achieve this have thusfar been disappointing. We aim to better define mechanisms of Treg development and function, to identify new ways of exploiting or blocking the suppressive function of Treg cells in individuals with inflammation and cancer (Figure 2).
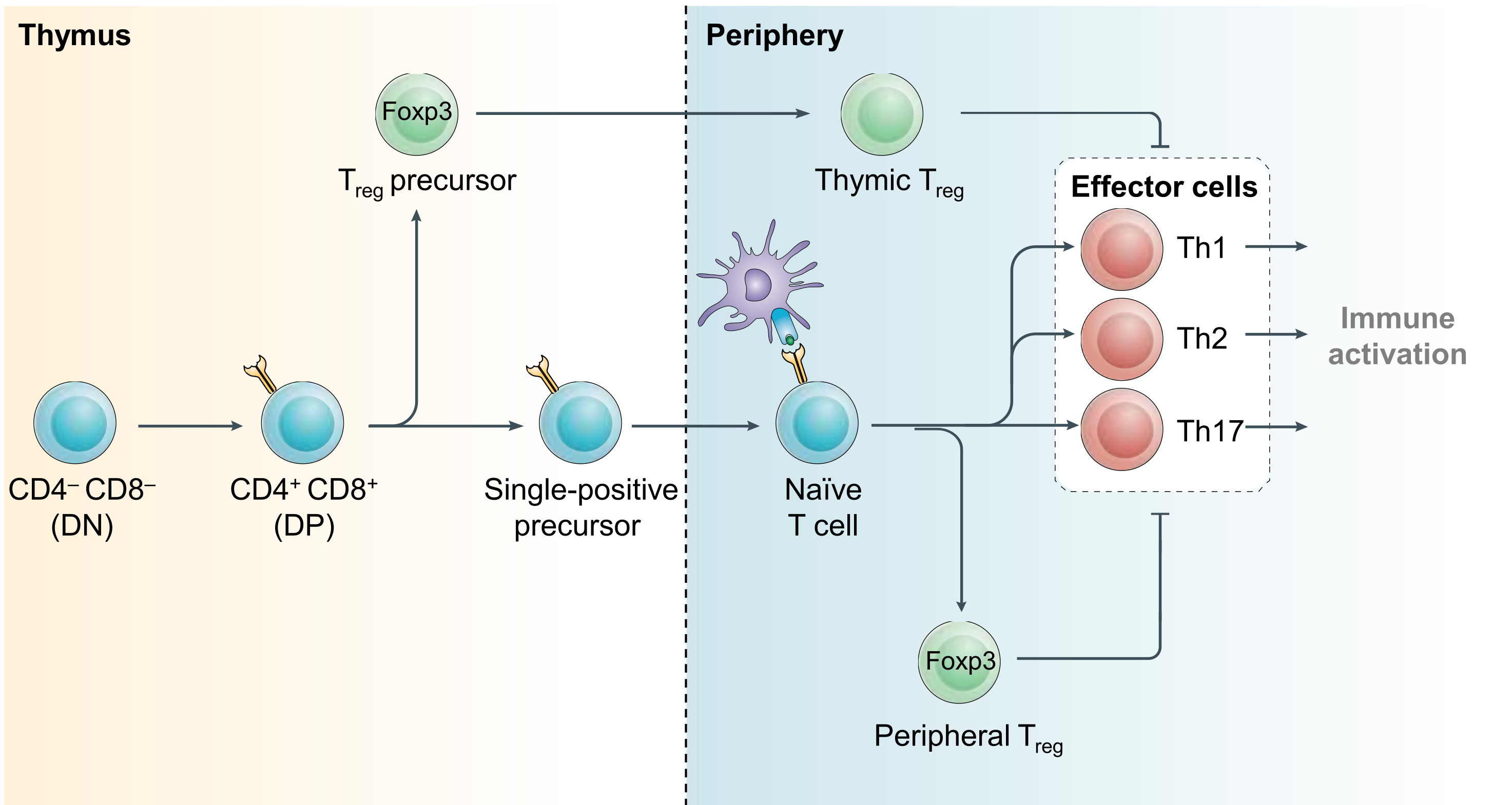
Figure 2. Regulatory T cell development. CD4+ effector and regulatory T (Treg) cells arise from common precursor cells within the thymus and periphery by exert opposing functions. Treg-mediated restraint of effector cell function is a critical immunoregulatory mechanism required to prevent lethal inflammation. Modified from Igarashi, Kurosaki and Roychoudhuri, Nat Rev Immunol 2017.
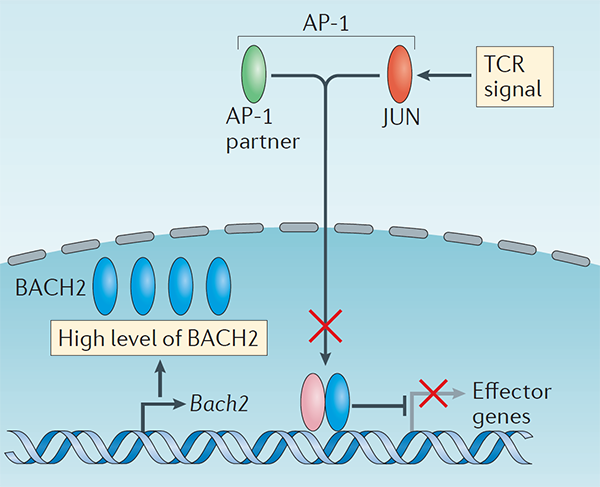
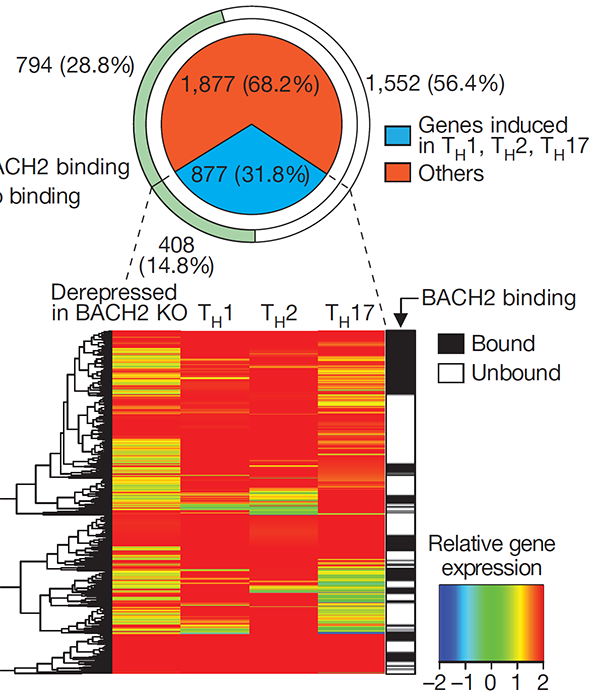
The immunoregulatory function of Treg cells is a major focus of the laboratory’s research. We have demonstrated the non-redundant requirement for the transcription factor BACH2 in Treg development (Nature 2013). Our findings provided a model of early Treg lineage commitment and explained why genetic polymorphisms at the human BACH2 locus are associated with autoimmune and allergic diseases. We established a now widely-accepted molecular model of how BACH2 functions in lymphocytes (Nat Immunol 2016; reviewed in Igarashi, Kurosaki and Roychoudhuri, Nat Rev Immunol 2017). Our research contributed to the discovery of a new human disease called BACH2-related Immunodeficiency and Autoimmunity (Afzali et al., Nat Immunol 2017). This has led to identification and improved management of patients with BRIDA. We showed that BACH2 promotes Treg-mediated cancer immunosuppression (J Clin Invest 2015). We have subsequently developed cell-based reporter assays for BACH2 function (Scientific Reports 2020), enabling a drug discovery programme in collaboration with Cancer Research UK Therapeutic Discovery Laboratories.
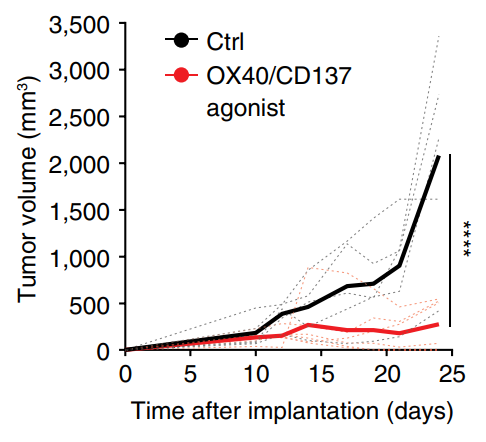
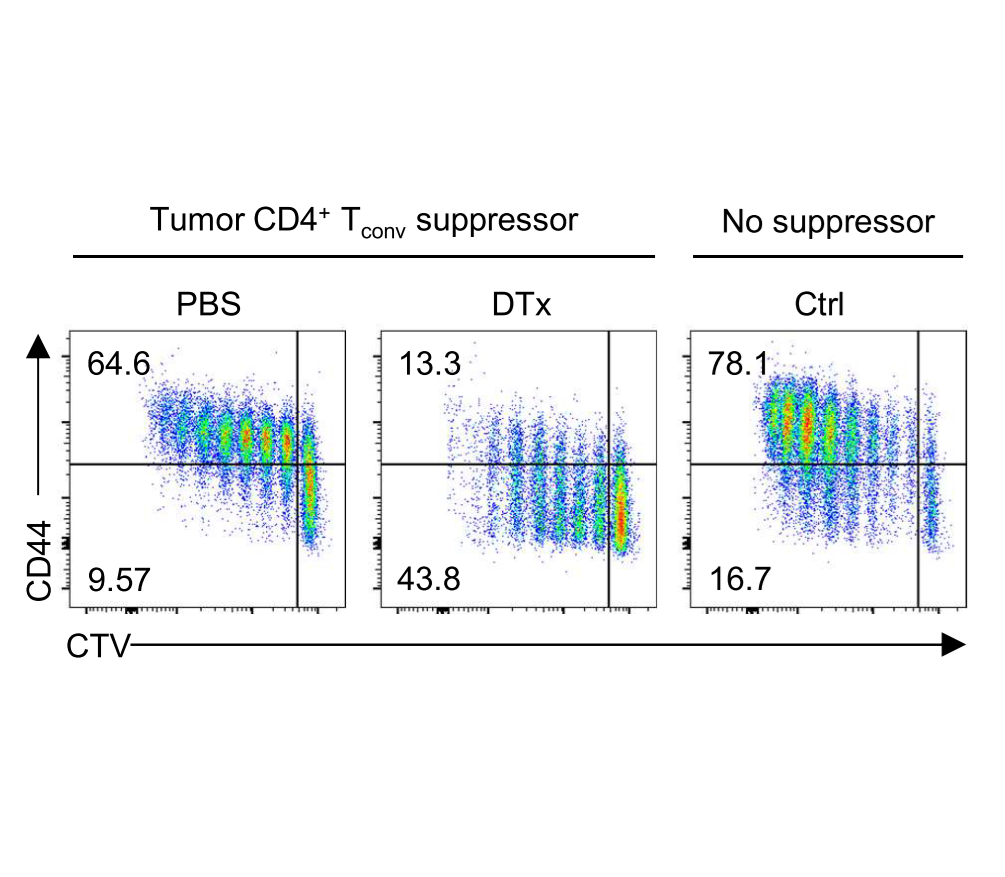
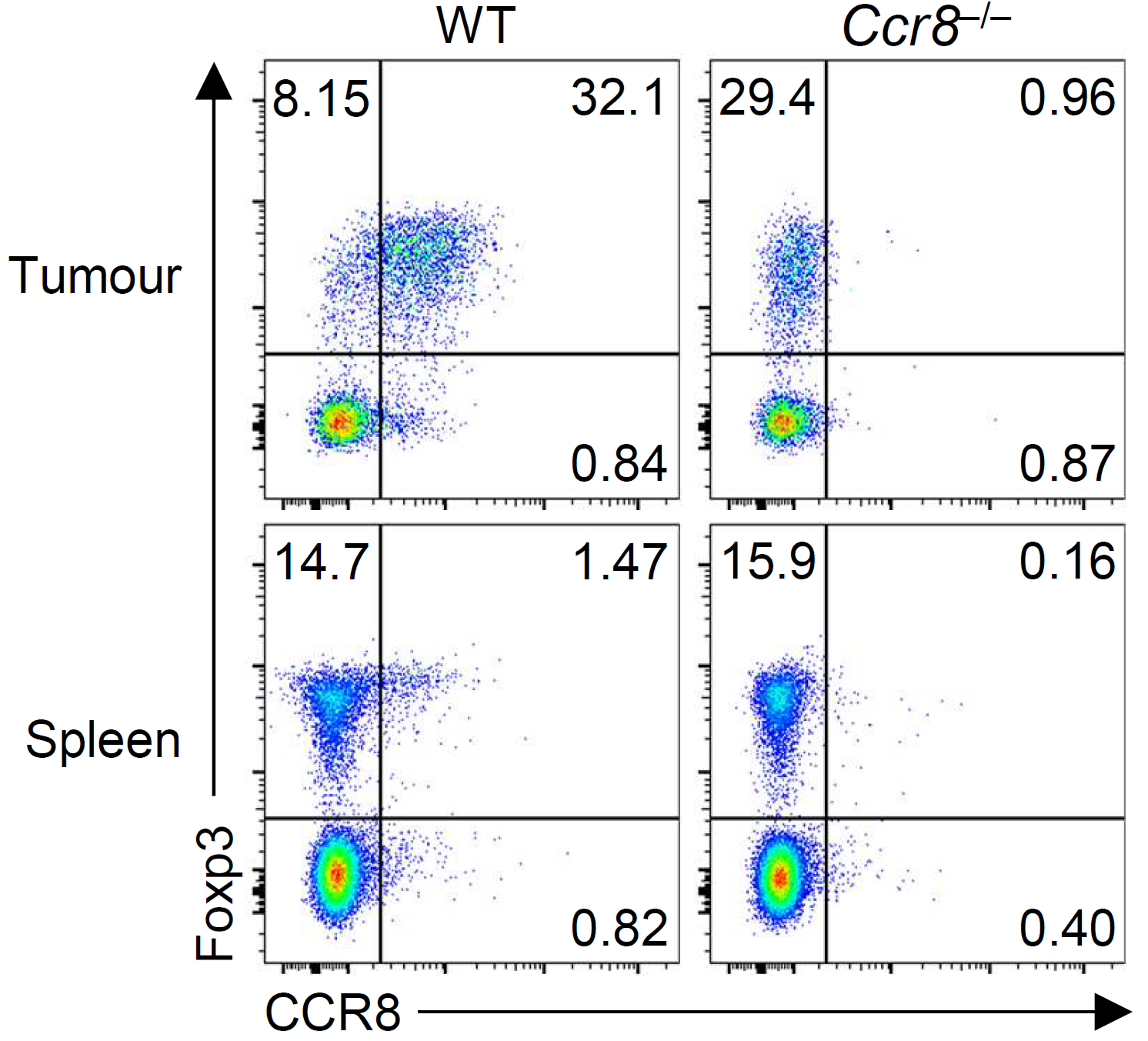
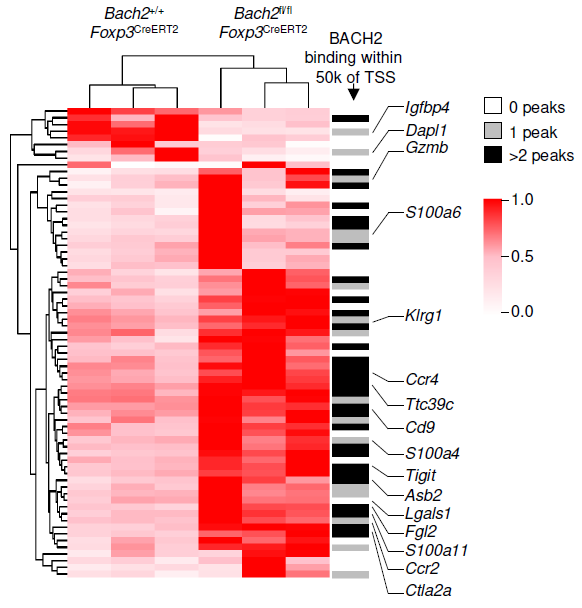
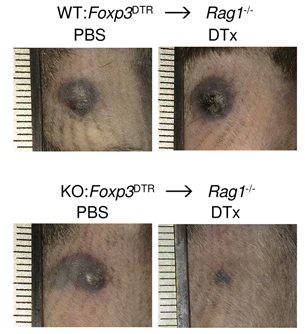
Our group showed that a distal enhancer at the prominent human autoimmune/allergic disease risk locus at chromosome 11q13.5 restricts gut inflammation by promoting expression of the TGF-b docking receptor GARP on Treg cells, revealing a novel mechanism of immune regulation in the gut (Nature 2020). We showed that quiescent cells marked by high levels of BACH2 expression are required for maintenance of Treg responses over time (Grant et al., J Exp Med 2020). We showed that CCR8 expression marks Treg cells with highly suppressive function in tumours but that it is dispensable for their accumulation and suppressive function (Immunology 2021). We have shown that CD4+ Tconv cells acquire counterregulatory suppressive functions upon therapeutic depletion of Treg cells, resulting in resistance to Treg-depleting therapies (Whiteside et al., Sci Immunol 2023). Our interest in targeting the immunosuppressive functions of Treg cells within tumours has led to a number of translational collaborations including an industrial collaboration with F-Star Therapeutics.
T cell maintenance and dysfunction
T cell responses are clonally expanded from small numbers of antigen-specific naive precursor cells which arose during thymic development. Upon priming, antigen-specific T cell responses must be maintained over long periods of time to enable T cell memory and durable responses to chronic antigens. Our laboratory is interested in the mechanisms that underpin long-lived T cell responses (Figure 3).
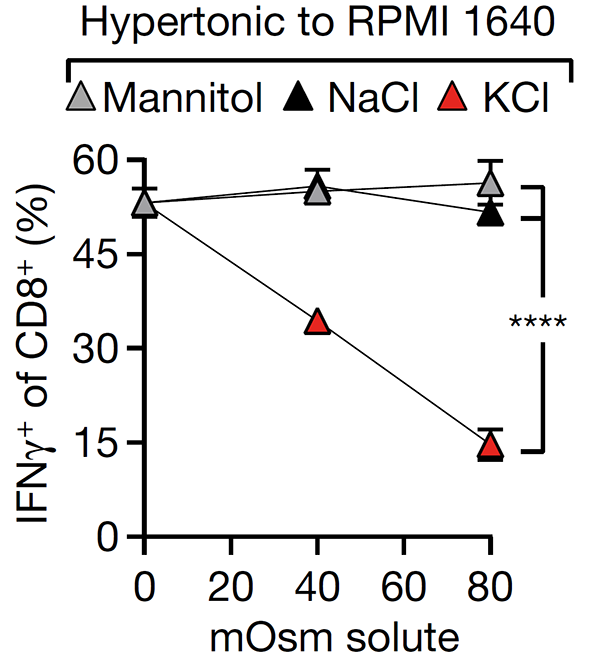
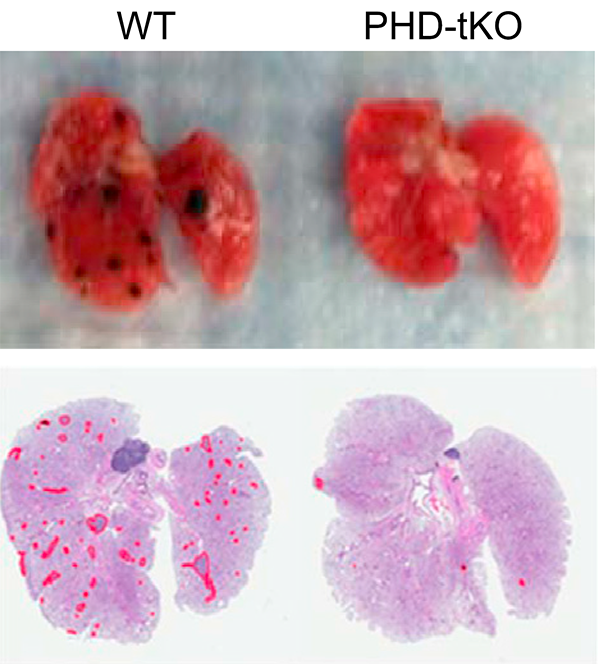
We have made progress in understanding how T cell dysfunction is induced by the interstitial microenvironment of cancer. We showed that high interstitial potassium concentrations within tumours limits CD8+ T cell function through suppression of the AKT/mTOR pathway (Nature 2016). We have shown that Treg differentiation is sensitive to local oxygen concentration contributing to lung immune homeostasis but creating a permissive environment for pulmonary cancer metastasis (Cell 2016). This research has led to new therapeutic opportunities and formed the basis for a number of industrial collaborations including with AstraZeneca and CRUK Therapeutic Discovery Laboratories.
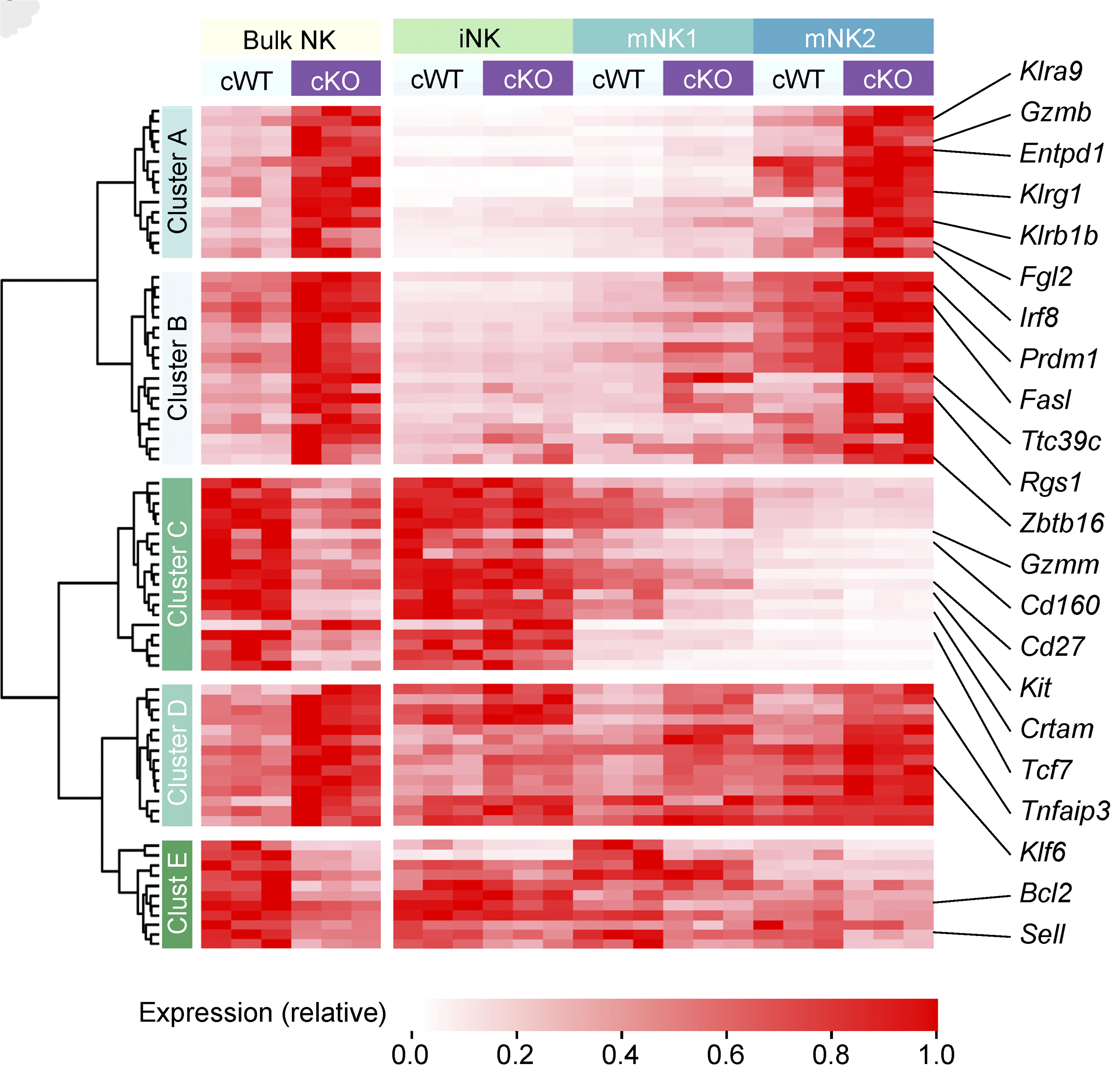
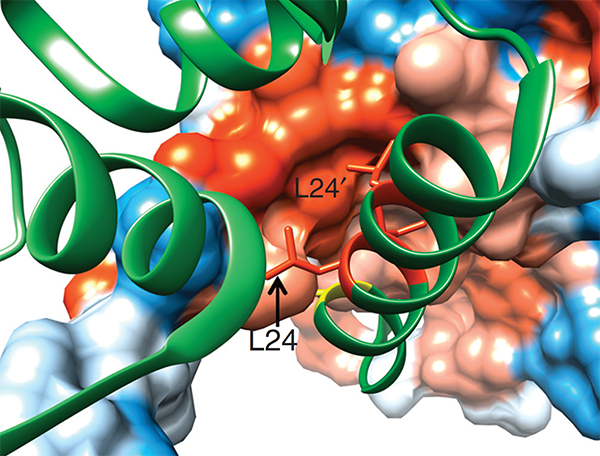
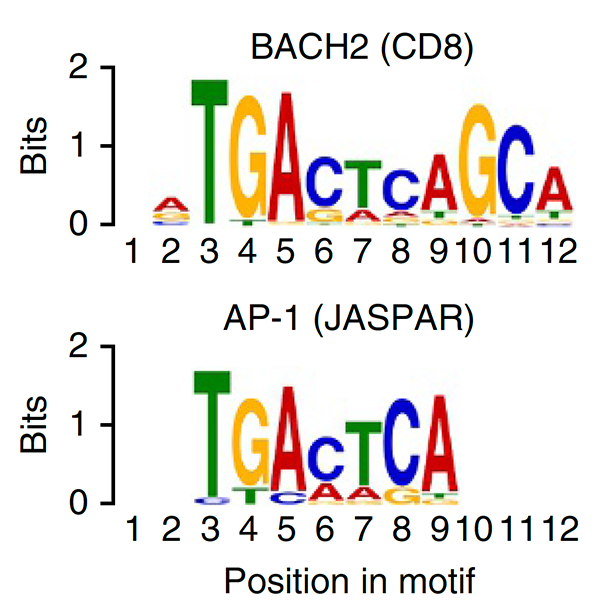
We have a long-standing interest in T cell maintenance. We conducted one of the earliest multiplexed single-cell gene expression analyses of immune cells revealing unappreciated heterogeneity in memory CD8+ T cell responses to vaccination (PNAS 2011). We defined transcriptional and epigenetic programmes of vaccine-induced memory T cells (Vaccine 2015, Cell Mol Immunol 2015). We showed that the transcription factor BACH2 functions as a quiescence factor promoting maintenance of long-lived memory CD8+ T cell responses to viral infection are maintained (Nat Immunol 2016). Mechanistically, BACH2 functions as a repressor of TCR-driven effector programmes by binding the genomic binding sites of AP-1 and sterically hindering access by AP-1 TFs. We showed that long-term maintenance of Treg populations is dependent upon the presence of a subset of functionally quiescent cells marked by high levels of Bach2 expression (J Exp Med 2020). We have also found that BACH2 functions as a quiescence factor in NK cells restricting NK-mediated immunosurveillance of lung metastasis (Imianowski et al., J Exp Med 2022). We have contributed to work showing that inhibition of AKT signalling enables expansion of T cells with a long-lived memory phenotype which mediates superior adoptive immunotherapy responses upon transfer into tumour-bearing recipients (Cancer Res, 2015), and that memory T cell–driven differentiation of naive cells impairs adoptive immunotherapy (J Clin Invest, 2015). Several projects in the laboratory now aim to exploit our understanding of T cell maintenance mechanisms to improve the long-term maintenance and function of T cells in the context of cell therapy, including CAR T cell and Treg cell therapy.
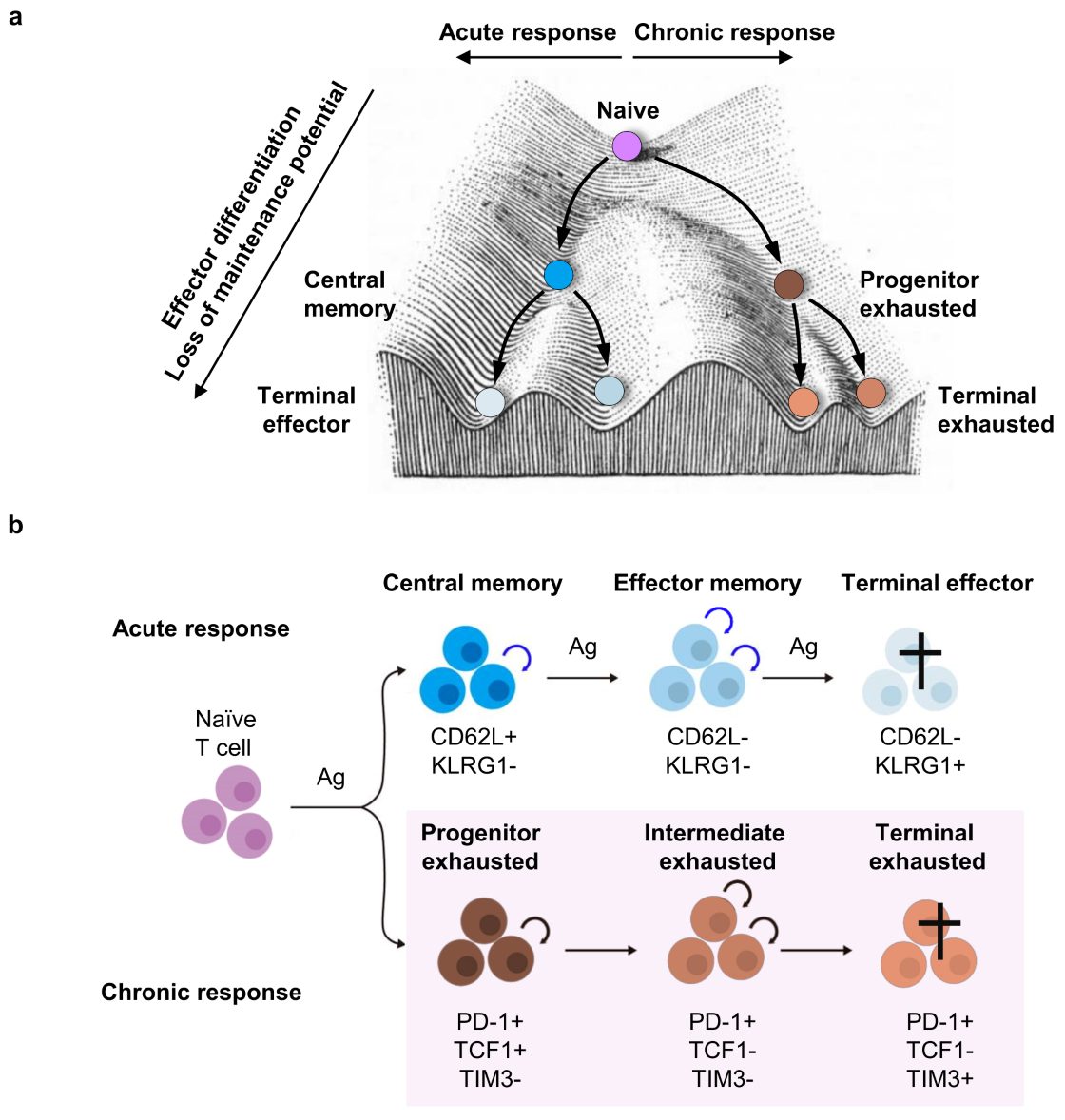
Figure 3. Maintenance of acute and chronic T cell responses. In both acute and chronic immune responses, T cells undergo progressive differentiation accompanied by acquisition of effector functions and loss of maintenance potential self-renewal and multipotency. This loss of maintenance potential is a key feature of the progression of T cells from naive, effector, and memory T cell subsets in acute responses, as well as progenitor, intermediate, and terminally exhausted T cells in chronic responses.
Research Highlights
(For a full list of publications see below)

Group Members
[Team] [Join us]@RoychoudhuriLab
Collaborators:
- Klaus Okkenhaug (Pathology)
- David Adams (Sanger)
- Adrian Liston (Babraham)
- Enrico Lugli (Humanitas, Milan)
- Tim Halim (CRUK CI)
- Suman Mitra (INSERM Lille)
- Gosia Trynka (Sanger)
Environment:
- Department of Pathology (Cambridge)
- CRUK Cambridge Centre (Cambridge)
- Babraham Institute (Cambridge)

